
Abstract
Multilocular cystic renal cell carcinoma is an uncommon low grade renal cell carcinoma with unique morphologic features. Its cytogenetic characteristics have not been fully investigated. Its relationship to typical clear cell renal cell carcinoma is uncertain. We evaluated 19 cases of multilocular cystic renal cell carcinoma diagnosed by strict morphologic criteria using the 2004 WHO classification system. The control group consisted of 19 low grade (Fuhrman grades 1 or 2) clear cell renal cell carcinomas. Chromosome 3p deletion status was determined by dual color interphase fluorescence in situ hybridization analysis. Chromosome 3p deletion was identified in 17 out of 19 (89%) of the clear cell renal cell carcinoma cases and 14 out of 19 (74%) of the multilocular cystic renal cell carcinoma cases, respectively. There was no difference in the status of chromosome 3p deletion between clear cell renal cell carcinoma and multilocular cystic renal cell carcinoma (P=0.40). These results support the concept that multilocular cystic renal cell carcinoma as a subtype of clear cell renal cell carcinoma.
Similar content being viewed by others
Main
Multilocular cystic renal cell carcinoma is an uncommon cystic variant of renal cell carcinoma with an excellent prognosis.1, 2, 3, 4, 5, 6 It is currently defined by strict morphologic criteria according to the 2004 World Health Organization (WHO) classification of tumors.1 Currently it is classified as a subtype of clear cell renal cell carcinoma; however, due to the excellent prognosis and distinct morphology debate exists regarding the nature of this tumor and whether it truly represents a variant of clear cell renal cell carcinoma.
Chromosome 3p deletion has been a consistent finding in clear cell renal cell carcinoma and is considered the hallmark cytogenetic abnormality found in clear cell renal cell carcinoma.7, 8, 9, 10 In this study we evaluated two-color interphase fluorescent in situ hybridization using unstained paraffin-embedded tissue slides to detect the presence or absence of 3p deletion. We studied samples from multilocular cystic renal cell carcinomas and compared the findings with those from a population of low-grade clear cell renal cell carcinomas.
Materials and methods
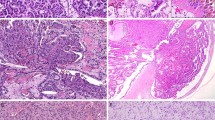
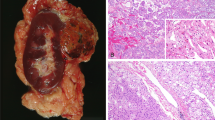
All 19 cases of multilocular cystic renal cell carcinoma strictly met the WHO criteria defining multilocular cystic renal cell carcinoma.1 These criteria include an entirely cystic tumor with thin septa containing clusters of Fuhrman grade 1 to 2 clear cells that do not expand the septa (Figure 1).1 None of these patients had von Hippel-Lindau syndrome or other genetic syndromes. Samples from 19 Fuhrman nuclear grade 1 and 2 clear cell renal cell carcinomas of the usual typical morphology were analyzed.

(a) Gross photograph of a partial nephrectomy specimen with multilocular cystic renal cell carcinoma showing an entirely cystic tumor with very thin septae that lack of expansile nodule. (b) Low power photomicrograph of the same tumor showing the complex cystic nature of the tumor that lack expansile nodules. (c and d) Higher power photomicrographs showing clear cells lining the septae. Also present are multiple small clusters of clear cells within the septae. No solid nodules are seen.
Fluorescence in situ hybridization (FISH) analyses were performed as described previously.11 Briefly, multiple 4 μm sections were obtained from formalin-fixed paraffin-embedded tissue blocks containing neoplastic tissue from each multilocular cystic renal cell carcinoma and clear cell renal cell carcinoma. A hematoxylin and eosin-stained slide from each block was examined to identify areas containing tumor cell clusters for cell counting. The remaining unstained slides were deparaffinized with two 15 min xylene washes. The slides were then washed twice with absolute ethanol, 10 min each, and then air-dried in the hood. FISH was performed with centromeric α-satellite DNA probes for 3 (CEP 3, Spectrum Orange), and subtelomeric probe for 3p25 (3pTel25, Spectrum Green). The probes were from Vysis (Downers Grove, IL) and were diluted with tDenHyb 2 (Insitus, Albuquerque, NM) in a ratio of 1:100. The slides were examined using a Zeiss Axioplan 2 microscope (ZEISS, Gottingen, Germany) with the following filters from Chroma (Chroma, Brattleboro, VT): SP-100 for 4′,6-diamidino-2-phenylindole, FITC MF-101 for Spectrum Green (3pTel25) and Gold 31003 for Spectrum Orange (CEP 3). The images were acquired with a charged coupled device camera and analyzed with MetaSystem Isis Software (MetaSystem, Belmont, MA). Four sequential focus stacks with 0.4-μm intervals were acquired and then integrated into a single image to reduce thickness-related artifacts. The method of analysis was partially described previously.12, 13, 14, 15, 16, 17, 18, 19, 20 In brief, for each slide, 100–150 nuclei from tumor tissue were scored for signals from probes under the fluorescence microscope with x1000 magnification. Nonneoplastic renal cortex was used as control tissue. The ratio of 3p/CEP3 signals was determined. The method to analyze 3p deletion was based on previous studies of deletion of chromosomes 1p and 19q in oligodendrogliomas.21 The cutoff value for 3p deletion was defined as a 3p/CEP3 ratio of <0.7, as previously described.11, 18, 19
Results
The patients with multilocular cystic renal cell carcinomas ranged in age from 33 to 72 years old (mean, 55 years) and those with clear cell renal cell carcinomas ranged in age from 42 to 74 years old (mean, 60 years) at the time of nephrectomy. The average diameter of the multilocular cystic renal cell carcinomas was 4.6 cm (range 1.5–12.9 cm) and it was 4.4 cm for the clear cell renal cell carcinomas (range 1.4–9.4 cm). The male to female ratio was 1.1:1 in the multilocular cystic renal cell carcinoma group and 0.9:1 in the clear cell renal cell carcinoma group. Fourteen of the multilocular cystic renal cell carcinomas were Fuhrman nuclear grade 1 (74%) and 5 were Fuhrman nuclear grade 2 (26%), whereas 2 cases of clear cell renal cell carcinoma were Fuhrman nuclear grade 1 (11%) and 17 cases were Fuhrman nuclear grade 2 (89%).
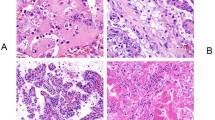
Nineteen cases of multilocular cystic renal cell carcinoma and nineteen cases of low grade clear cell renal cell carcinoma were investigated for chromosome 3p deletion. All of the slides showed well defined hybridization signals. Fourteen of the 19 multilocular cystic renal cell carcinomas were shown to have chromosome 3p deletion (74%) by FISH analysis. Seventeen of the 19 clear cell renal cell carcinomas were shown to have chromosome 3p deletion (89%) (Figure 2). The difference in the status of chromosome 3p deletion between the groups was not statistically significant (P=0.4).

Multilocular cystic renal cell carcinoma. (a) Low power photomicrograph shows the predominantly multilocular architecture with variably sized cysts and thin septae. (b) The septae are lined by clear cells without a solid component. (c) Clusters of neoplastic clear cells are seen within the septae. The nuclei are Fuhrman grade 1. (d) Fluorescence in situ hybridization (FISH) shows tumor nuclei with 2 orange signals (CEP3) and one green signal (Tel 3p25) demonstrating 3p deletion.
Discussion
Multilocular cystic renal cell carcinoma, also known as multilocular cystic clear cell renal cell carcinoma and multicystic clear cell carcinoma, is a rare variant of renal cell carcinoma.1, 22 Multilocular cystic renal cell carcinoma is almost always discovered incidentally before the onset of symptoms.23 The differential diagnosis of this entity consists of other cystic lesions of the kidney, primarily cystic nephroma, extensively cystic clear cell renal cell carcinoma, clear cell papillary renal cell carcinoma, and tubulocystic carcinoma. While cystic nephroma may have at least some clear cells lining the septa, the lining clear cells tend to be focally rather than diffusely distributed, and there are no clusters of clear cells in the walls. The ovarian-like stroma in cystic nephroma, if present, distinguishes it from multilocular cystic renal cell carcinoma, which does not exhibit this type of stroma. Extensively cystic clear cell renal cell carcinoma is distinguished from multilocular cystic renal cell carcinoma by the fact that solid areas may be evident grossly, or expansile nodules of clear cells are observed microscopically. Clear cell papillary renal cell carcinoma is usually cystic and the cyst walls are lined by clear cells; however, much of the tumor exhibits papillary architecture, a feature not found in multilocular cystic renal cell carcinoma.18, 24 Tubulocystic carcinoma is another multicystic tumor that has been increasingly recognized in the past 15 years.25 It differs from multilocular cystic renal cell carcinoma in that the cystic spaces, rather than being lined by clear cells, are lined by flat cuboidal and sometimes hobnail-type cells with eosinophilic cytoplasm and variable nuclear atypia, typically with nucleolar prominence in the range of Fuhrman grade 2 or 3, another feature incompatible with a diagnosis of multilocular cystic renal cell carcinoma. In addition, the septal structures of tubulocystic carcinoma do not harbor clusters of clear cells.
In this series, the average size of the multilocular cystic renal cell carcinomas (4.6 cm) is similar to that of typical low-grade clear cell renal cell carcinomas (4.4 cm). Multilocular cystic renal cell carcinoma is virtually always confined to the kidney when diagnosed and hence is at low stage, and by definition has a low Fuhrman grade. Even though multilocular cystic renal cell carcinoma is a histologically distinct lesion, little is known about its molecular genetic characteristics. Deletions in chromosome 3p are a consistent finding in clear cell renal cell carcinoma. These mutations are considered to be one of the primary events in the pathogenesis of this carcinoma. These losses have been reported in 70–90% of clear cell renal cell carcinomas and are rarely seen in other types of renal cell carcinoma.11, 26, 27 One of the genes shown to be involved in the development of clear cell renal cell carcinoma is the von Hippel-Lindau (VHL) tumor suppressor gene located at the 3p25 chromosomal region.28, 29 It is consistently inactivated in both sporadic and hereditary clear cell renal cell carcinoma, most commonly by mutation of one allele followed by deletion of another allele, or less commonly by VHL gene mutations or promoter hypermethylation. In the study including 177 clear cell renal cell carcinomas, Young and colleagues found 3p deletion in 89% of the cases by LOH, VHL mutation in 75%, and methylation in 31% of evaluable tumors. Only 3% of the cases showed wild type VHL.30
In this study we performed FISH analysis for 3p deletion on samples from multilocular cystic renal cell carcinomas and compared the findings with those from a population of similarly low grade conventional clear cell renal cell carcinomas. Deletion in 3p was observed in 74% of the multilocular cystic renal cell carcinomas and in 89% of the clear cell renal cell carcinomas with no statistically significant difference in the incidence of 3p deletion, between the groups. Tumors that develop through carcinogenesis pathways other than VHL gene deletion may not be detectable by using FISH methods, and this may account for some of the tumors that were negative for the 3p deletion. Our findings support the hypothesis that multilocular cystic renal cell carcinoma is a subtype of clear cell renal cell carcinoma. Using a strict definition and diagnostic criteria, it appears that multilocular cystic renal cell carcinomas represent a homogeneous entity both clinically and genetically.
The VHL gene plays an important role in carcinogenesis but not in the progression of clear cell renal cell carcinoma, since its loss appears to have no influence upon the grade or stage of tumors.31 Regions of chromosome 3p other than the site harboring the VHL gene (3p25) have been implicated in the progression of clear cell renal cell carcinoma and have been shown to harbor additional tumor suppressor genes.8 In addition to DNA alterations on chromosome 3, other chromosomal abnormalities such as losses of chromosome arms 14q, 9p, 8p and 6q have been identified in clear cell renal cell carcinoma, some of which have been shown to have a close correlation with higher stage and a worse outcome.7, 32, 33, 34, 35, 36, 37, 38, 39 It remains unclear whether other chromosomal or gene alterations play a role in the pathogenesis or progression of multilocular cystic renal cell carcinoma. The fact that additional mutations and chromosomal alterations are frequently present in high grade clear cell renal cell carcinomas, but are rarely reported in multilocular renal cell carcinoma, might explain the excellent prognosis of multilocular cystic renal cell carcinoma. Additional molecular characterization of these tumors is needed to provide more insight into the pathogenesis of this entity as well as further clarification of predictors of outcome.
Our observation of chromosome 3p deletion in multilocular cystic renal cell carcinoma provides support for the hypothesis that it is a subtype of clear cell renal cell carcinoma; however, we believe that usage of the term ‘multilocular cystic renal cell carcinoma’ should be continued in practice, since it denotes a morphologically distinctive tumor that is cured by resection in essentially 100% of cases.
References
Eble JN . Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs. IARC Press, Oxford University Press (distributor): Lyon, Oxford, 2004.
Halat SK, MacLennan GT . Multilocular cystic renal cell carcinoma. J Urol 2007;177:343.
Lopez-Beltran A, Scarpelli M, Montironi R, et al. 2004 WHO classification of the renal tumors of the adults. Eur Urol 2006;49:798–805.
Murphy WM, Grignon DJ, Perlman EJ . Tumors of the Kidney, Bladder, and Related Urinary Structures. American Registry of Pathology: Washington, DC, 2004.
Nassir A, Jollimore J, Gupta R, et al. Multilocular cystic renal cell carcinoma: a series of 12 cases and review of the literature. Urology 2002;60:421–427.
Suzigan S, Lopez-Beltran A, Montironi R, et al. Multilocular cystic renal cell carcinoma : a report of 45 cases of a kidney tumor of low malignant potential. Am J Clin Pathol 2006;125:217–222.
Cheng L, Zhang S, MacLennan GT, et al. Molecular and cytogenetic insights into the pathogenesis, classification, differential diagnosis, and prognosis of renal epithelial neoplasms. Hum Pathol 2009;40:10–29.
Cohen HT, McGovern FJ . Renal-cell carcinoma. N Engl J Med 2005;353:2477–2490.
Jones TD, Eble JN, Cheng L . Application of molecular diagnostic techniques to renal epithelial neoplasms. Clin Lab Med 2005;25:279–303.
Kovacs G, Erlandsson R, Boldog F, et al. Consistent chromosome 3p deletion and loss of heterozygosity in renal cell carcinoma. Proc Natl Acad Sci USA 1988;85:1571–1575.
Cheng L, MacLennan GT, Zhang S, et al. Evidence for polyclonal origin of multifocal clear cell renal cell carcinoma. Clin Cancer Res 2008;14:8087–8093.
Brunelli M, Eble JN, Zhang S, et al. Gains of chromosomes 7, 17, 12, 16, and 20 and loss of Y occur early in the evolution of papillary renal cell neoplasia: a fluorescent in situ hybridization study. Mod Pathol 2003;16:1053–1059.
Brunelli M, Eccher A, Gobbo S, et al. Loss of chromosome 9p is an independent prognostic factor in patients with clear cell renal cell carcinoma. Mod Pathol 2008;21:1–6.
Brunelli M, Gobbo S, Cossu-Rocca P, et al. Chromosomal gains in the sarcomatoid transformation of chromophobe renal cell carcinoma. Mod Pathol 2007;20:303–309.
Cheng L, Zhang S, MacLennan GT, et al. Interphase fluorescence in situ hybridization analysis of chromosome 12p abnormalities is useful for distinguishing epidermoid cysts of the testis from pure mature teratoma. Clin Cancer Res 2006;12:5668–5672.
Cossu-Rocca P, Eble JN, Delahunt B, et al. Renal mucinous tubular and spindle carcinoma lacks the gains of chromosomes 7 and 17 and losses of chromosome Y that are prevalent in papillary renal cell carcinoma. Mod Pathol 2006;19:488–493.
Cossu-Rocca P, Eble JN, Zhang S, et al. Acquired cystic disease-associated renal tumors: an immunohistochemical and fluorescence in situ hybridization study. Mod Pathol 2006;19:780–787.
Gobbo S, Eble JN, Grignon DJ, et al. Clear cell papillary renal cell carcinoma: a distinct histopathologic and molecular genetic entity. Am J Surg Pathol 2008;32:1239–1245.
Gobbo S, Eble JN, Maclennan GT, et al. Renal cell carcinomas with papillary architecture and clear cell components: the utility of immunohistochemical and cytogenetical analyses in differential diagnosis. Am J Surg Pathol 2008;32:1780–1786.
Jones TD, Eble JN, Wang M, et al. Clonal divergence and genetic heterogeneity in clear cell renal cell carcinomas with sarcomatoid transformation. Cancer 2005;104:1195–1203.
Fallon KB, Palmer CA, Roth KA, et al. Prognostic value of 1p, 19q, 9p, 10q, and EGFR-FISH analyses in recurrent oligodendrogliomas. J Neuropathol Exp Neurol 2004;63:314–322.
Eble JN, Bonsib SM . Extensively cystic renal neoplasms: cystic nephroma, cystic partially differentiated nephroblastoma, multilocular cystic renal cell carcinoma, and cystic hamartoma of renal pelvis. Semin Diagn Pathol 1998;15:2–20.
Corica FA, Iczkowski KA, Cheng L, et al. Cystic renal cell carcinoma is cured by resection: a study of 24 cases with long-term followup. J Urol 1999;161:408–411.
Gobbo S, Eble JN, MacLennan GT, et al. Renal cell carcinomas with papillary architecture and clear cell components: the utility of immunohistochemical and cytogenetical analyses in differential diagnosis. Am J Surg Pathol 2008;32:1780–1786.
Amin MB, MacLennan GT, Gupta R, et al. Tubulocystic carcinoma of the kidney: clinicopathologic analysis of 31 cases of a distinctive rare subtype of renal cell carcinoma. Am J Surg Pathol 2009;33:384–392.
Barocas DA, Mathew S, DelPizzo JJ, et al. Renal cell carcinoma sub-typing by histopathology and fluorescence in situ hybridization on a needle-biopsy specimen. BJU international 2007;99:290–295.
Sanjmyatav J, Rubtsov N, Starke H, et al. Identification of tumor entities of renal cell carcinoma using interphase fluorescence in situ hybridization. The Journal of Urology 2005;174:731–735.
Iliopoulos O, Eng C . Genetic and clinical aspects of familial renal neoplasms. Semin Oncol 2000;27:138–149.
Wagner JR, Linehan WM . Molecular genetics of renal cell carcinoma. Semin Urol Oncol 1996;14:244–249.
Young AC, Craven RA, Cohen D, et al. Analysis of VHL Gene alterations and their relationship to clinical parameters in sporadic conventional renal cell carcinoma. Clin Cancer Res 2009;15:7582–7592.
Wu SQ, Hafez GR, Xing W, et al. The correlation between the loss of chromosome 14q with histologic tumor grade, pathologic stage, and outcome of patients with nonpapillary renal cell carcinoma. Cancer 1996;77:1154–1160.
Beroud C, Fournet JC, Jeanpierre C, et al. Correlations of allelic imbalance of chromosome 14 with adverse prognostic parameters in 148 renal cell carcinomas. Genes Chromosomes Cancer 1996;17:215–224.
Cairns P, Tokino K, Eby Y, et al. Localization of tumor suppressor loci on chromosome 9 in primary human renal cell carcinomas. Cancer Res 1995;55:224–227.
Grady B, Goharderakhshan R, Chang J, et al. Frequently deleted loci on chromosome 9 may harbor several tumor suppressor genes in human renal cell carcinoma. J Urol 2001;166:1088–1092.
Kovacs G, Frisch S . Clonal chromosome abnormalities in tumor cells from patients with sporadic renal cell carcinomas. Cancer Res 1989;49:651–659.
Mitsumori K, Kittleson JM, Itoh N, et al. Chromosome 14q LOH in localized clear cell renal cell carcinoma. J Pathol 2002;198:110–114.
Moch H, Presti Jr JC, Sauter G, et al. Genetic aberrations detected by comparative genomic hybridization are associated with clinical outcome in renal cell carcinoma. Cancer Res 1996;56:27–30.
Presti Jr JC, Rao PH, Chen Q, et al. Histopathological, cytogenetic, and molecular characterization of renal cortical tumors. Cancer Res 1991;51:1544–1552.
Schullerus D, Herbers J, Chudek J, et al. Loss of heterozygosity at chromosomes 8p, 9p, and 14q is associated with stage and grade of non-papillary renal cell carcinomas. J Pathol 1997;183:151–155.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Halat, S., Eble, J., Grignon, D. et al. Multilocular cystic renal cell carcinoma is a subtype of clear cell renal cell carcinoma. Mod Pathol 23, 931–936 (2010). https://doi.org/10.1038/modpathol.2010.78
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2010.78
Keywords
This article is cited by
-
Clear cell renal cell carcinoma with cystic component similar to multilocular cystic renal neoplasm of low malignant potential: a rare pattern of cyst-dependent progression from multilocular cystic renal neoplasm of low malignant potential
Diagnostic Pathology (2023)
-
Comparison of survival between unilocular cystic and purely solid renal cell carcinoma
Scientific Reports (2022)
-
Histologische Subtypen des Nierenzellkarzinoms
Der Pathologe (2021)
-
Pathologie und Molekularpathologie des Nierenzellkarzinoms
Der Onkologe (2019)
-
Predominantly cystic clear cell renal cell carcinoma and multilocular cystic renal neoplasm of low malignant potential form a low-grade spectrum
Virchows Archiv (2018)
